Classify samples as being either SARS-CoV-2 positive or negative, identify the strain of virus, and produce statistics about the mapping.
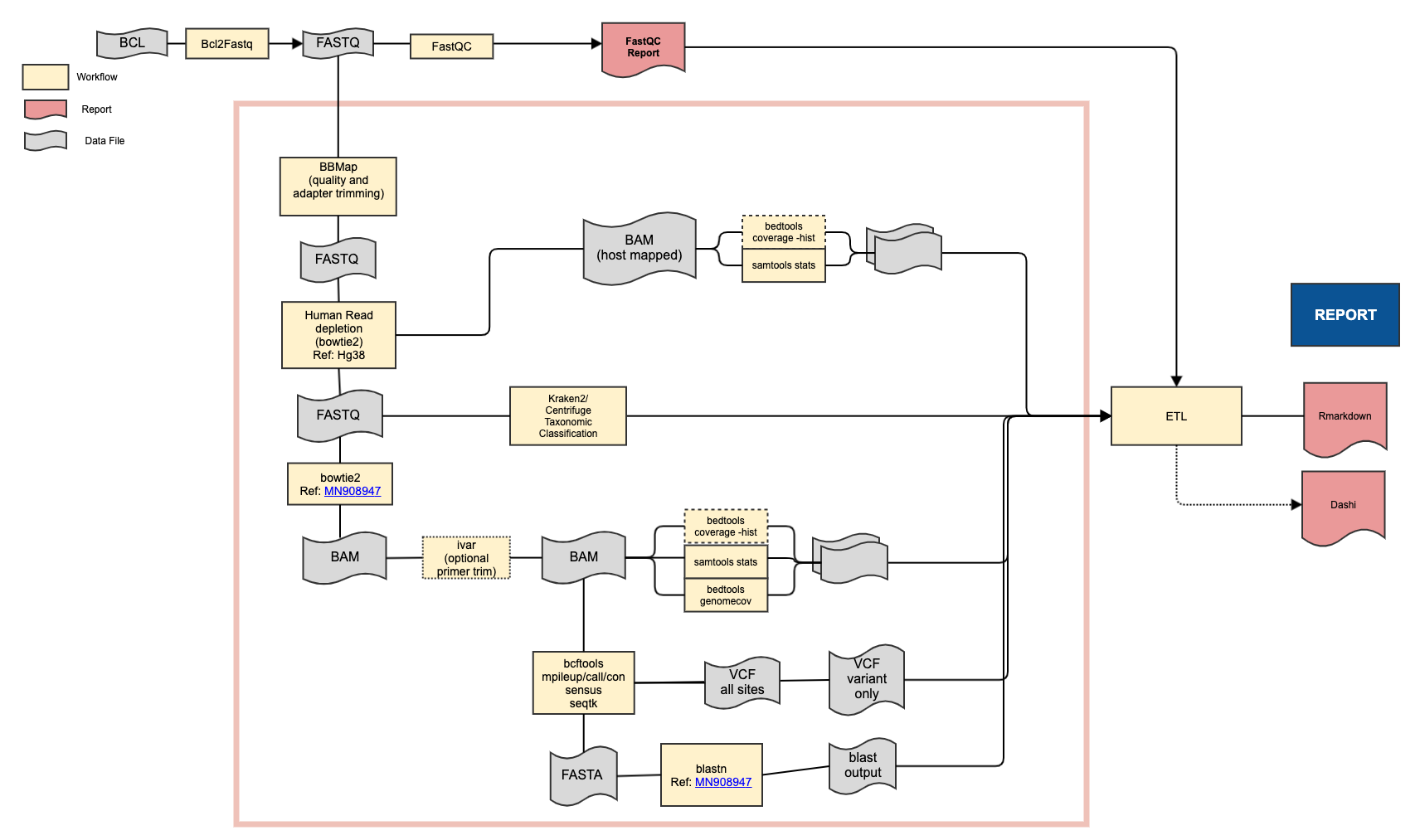
- bbmap 38.75
- bowtie2 2.3.5.1
- samtools 1.9
- kraken2 2.0.8
- ivar 1.0
- bcftools 1.9
- vcftools 0.1.16
- seqtk 1.3
- bedtools 2.27
- blast 2.8.1
- spades 3.14.0
java -jar cromwell.jar run sarsCoV2Analysis.wdl --inputs inputs.json
| Parameter | Value | Description |
|---|---|---|
fastq1 |
File | Read 1 fastq file, gzipped. Can be either targeted or whole transcriptome |
fastq2 |
File | Read 2 fastq file, gzipped. Can be either targeted or whole transcriptome. |
samplePrefix |
String | Prefix for output files |
| Parameter | Value | Default | Description |
|---|---|---|---|
primerBed |
String? | None | bed file used to trim the primers off of the bam sequences |
panelBed |
String? | None | bed file for an optional Panel of Intervals |
readCount |
Int? | None | Optionally pass the number of reads in the input fastq files |
doAssembly |
Boolean | false | Flag to control building an assembly with SPADES |
| Parameter | Value | Default | Description |
|---|---|---|---|
bbMap.modules |
String | "bbmap/38.75" | Modules to load for the task |
bbMap.reference |
String | "$BBMAP_ROOT/share/bbmap/resources/adapters.fa" | Reference FSATA, adapter sequences |
bbMap.trimq |
Int | 25 | Trim quality |
bbMap.mem |
Int | 8 | Memory allocated to the task |
bbMap.timeout |
Int | 72 | Timeout, in hours |
bowtie2HumanDepletion.modules |
String | "bowtie2/2.3.5.1 samtools/1.9 hg38-bowtie-index/2.3.5.1" | Modules to load for the task |
bowtie2HumanDepletion.reference |
String | "$HG38_BOWTIE_INDEX_ROOT/hg38_random_index" | Reference FSATA, adapter sequences |
bowtie2HumanDepletion.mem |
Int | 12 | Memory allocated to the task |
bowtie2HumanDepletion.timeout |
Int | 72 | Timeout, in hours |
bowtie2HumanDepletion.threads |
Int | 8 | Threads to use for this task |
kraken2.modules |
String | "kraken2/2.0.8 kraken2-database/1" | Modules to load for the task |
kraken2.kraken2DB |
String | "$KRAKEN2_DATABASE_ROOT/" | Root of the directory with KRAKEN database |
kraken2.mem |
Int | 8 | Memory allocated to the task |
kraken2.timeout |
Int | 72 | Timeout, in hours |
bowtie2Sensitive.modules |
String | "bowtie2/2.3.5.1 sars-covid-2-polymasked-bowtie-index/2.3.5.1 samtools/1.9" | Modules to load for the task |
bowtie2Sensitive.sarsCovidIndex |
String | "$SARS_COVID_2_POLYMASKED_BOWTIE_INDEX_ROOT/MN908947.3.mask" | Polymasked Bowtie2 index file |
bowtie2Sensitive.mem |
Int | 8 | Memory allocated to the task |
bowtie2Sensitive.timeout |
Int | 72 | Timeout, in hours |
bowtie2Sensitive.threads |
Int | 4 | Threads to use for the task |
articTrimming.modules |
String | "ivar/1.0 bedtools" | Environment module name and version to load (space separated) before command execution. |
articTrimming.allowNoprimer |
Boolean? | None | Allow reads that don't have primer sequence? Ligation prep = false, nextera = true. |
articTrimming.mem |
Int | 8 | Memory (in GB) to allocate to the job. |
articTrimming.timeout |
Int | 72 | Maximum amount of time (in hours) the task can run for. |
variantCalling.modules |
String | "bcftools/1.9 samtools/1.9 vcftools/0.1.16 seqtk/1.3 sars-covid-2-polymasked/mn908947.3" | Modules to load for the task |
variantCalling.sarsCovidRef |
String | "$SARS_COVID_2_POLYMASKED_ROOT/MN908947.3.mask.fasta" | Path to sarsCovidRef reference file |
variantCalling.mem |
Int | 8 | Memory allocated to the task |
variantCalling.timeout |
Int | 72 | Timeout, in hours |
qcStats.modules |
String | "bedtools samtools/1.9" | Modules to load for the task |
qcStats.mem |
Int | 8 | Memory allocated to the task |
qcStats.timeout |
Int | 72 | Timeout, in hours |
blast2ReferenceSequence.modules |
String | "blast sars-covid-2-polymasked/mn908947.3" | Modules to load for the task |
blast2ReferenceSequence.reference |
String | "$SARS_COVID_2_POLYMASKED_ROOT/MN908947.3.mask.fasta" | Reference FASTA file |
blast2ReferenceSequence.mem |
Int | 8 | Memory allocated to the task |
blast2ReferenceSequence.timeout |
Int | 72 | Timeout, in hours |
generateReadCount.modules |
String | "" | Modules to load for the task |
generateReadCount.mem |
Int | 4 | Memory allocated to the task |
generateReadCount.timeout |
Int | 4 | Timeout, in hours |
spadesGenomicAssembly.modules |
String | "spades/3.14.0" | Modules to load for the task |
spadesGenomicAssembly.minReads |
Int | 100 | threshold for minimum reads |
spadesGenomicAssembly.mem |
Int | 8 | Memory allocated to the task |
spadesGenomicAssembly.timeout |
Int | 72 | Timeout, in hours |
| Output | Type | Description | Labels |
|---|---|---|---|
hostRemovedR1Fastq |
File | Fastq file R1 with host reads removed. | vidarr_label: hostRemovedR1Fastq |
hostRemovedR2Fastq |
File | Fastq file R2 with host reads removed. | vidarr_label: hostRemovedR2Fastq |
hostMappedBam |
File | Reads mapped to host, bam format. | vidarr_label: hostMappedBam |
hostMappedBai |
File | Index for bam with host-mapped reads. | vidarr_label: hostMappedBai |
taxonomicClassification |
File | Kraken2 taxonomic classification report. | vidarr_label: taxonomicClassification |
bam |
File | Bowtie2-aligned reads, sensisitve mode. | vidarr_label: bam |
bai |
File | Index of bam with bowtie2-aligned reads, sensisitve mode. | vidarr_label: bai |
primertrimSortedBam |
File? | Trimmed reads aligned with bowtie2. | vidarr_label: primertrimSortedBam |
primertrimSortedBai |
File? | Index for trimmed reads aligned with bowtie2. | vidarr_label: primertrimSortedBai |
vcf |
File | Variants produced with bcftools (using cleaned reads). | vidarr_label: vcf |
consensusFasta |
File | Consensus fasta file produced along the variants. | vidarr_label: consensusFasta |
variantOnlyVcf |
File | Variants produced with bcftools, only variant calls. | vidarr_label: variantOnlyVcf |
bl2seqReport |
File | BLAST results. | vidarr_label: bl2seqReport |
cvgHist |
File? | Coverage histogram, output from qcstats. | vidarr_label: cvgHist |
genomecvgHist |
File | Genome coverage histogram. | vidarr_label: genomecvgHist |
genomecvgPerBase |
File | Genome coverage per base. | vidarr_label: genomecvgPerBase |
hostMappedAlignmentStats |
File | Stats for host-aligned reads. | vidarr_label: hostMappedAlignmentStats |
hostDepletedAlignmentStats |
File | Stats for alignments of reads depleted of host. | vidarr_label: hostDepletedAlignmentStats |
spades |
File? | Spades output. | vidarr_label: spades |
This section lists command(s) run by sarsCoV2Analysis workflow
- Running sarsCoV2Analysis
set -euo pipefail
#Remove adapters and quality trim
bbmap bbduk in1=~{fastq1} in2=~{fastq2} \
out1=~{sample}_qad_r1.fastq.gz out2=~{sample}_qad_r2.fastq.gz \
ref=~{reference} \
ktrim=r k=23 mink=11 hdist=1 tpe tbo qtrim=rl trimq=~{trimq}
set -euo pipefail
#Align fastq files to hg38 & only keep unmapped
bowtie2 -p ~{threads} -x ~{reference} \
-1 ~{fastq1} -2 ~{fastq2} \
--un-conc-gz ~{sample}_host_removed_r%.fastq.gz | \
samtools view -b | \
samtools sort - -o ~{hostMappedBam_}
samtools index ~{hostMappedBam_}
set -euo pipefail
kraken2 --paired ~{fastq1} ~{fastq2} \
--db ~{kraken2DB} \
--report ~{sample}.kreport2.txt
set -euo pipefail
#Align reads to reference
bowtie2 --sensitive-local -p ~{threads} -x ~{sarsCovidIndex} \
-1 ~{fastq1} -2 ~{fastq2} | samtools view -b | samtools sort - -o ~{bamFile_}
samtools index ~{bamFile_}
set -euo pipefail
ivar trim -i ~{bam} -b ~{primerBed} -p ~{primertrim} ~{true="-e" false="" allowNoprimer}
samtools sort ~{primertrimBam} -o ~{sortedBam_}
samtools index ~{sortedBam_}
set -euo pipefail
#Call consensus sequence
samtools mpileup -aa -uf ~{sarsCovidRef} ~{bam} | \
bcftools call --ploidy 1 -Mc | tee -a ~{vcfName} | \
vcfutils.pl vcf2fq -d 10 | \
seqtk seq -A - | sed '2~2s/[actg]/N/g' > ~{fastaName}
bcftools mpileup -a "INFO/AD,FORMAT/DP,FORMAT/AD" \
-d 8000 -f ~{sarsCovidRef} ~{bam} | \
tee ~{sample}.m.vcf | bcftools call --ploidy 1 -m -v > ~{variantOnlyVcf_}
set -euo pipefail
if [ -f "~{panelBed}" ]; then
bedtools coverage -hist -a ~{panelBed} -b ~{bam} > ~{sample}.cvghist.txt
fi
bedtools genomecov -ibam ~{bam} > ~{sample}.genomecvghist.txt
bedtools genomecov -d -ibam ~{bam} > ~{sample}.genome.cvgperbase.txt
samtools stats ~{hostMappedBam} > ~{sample}.host.mapped.samstats.txt
samtools stats ~{bam} > ~{sample}.samstats.txt
set -euo pipefail
# Suppress error for negative controls or samples with very little reads
if blastn -query ~{consensusFasta} -subject ~{reference} \
-word_size 28 -reward 1 -penalty -2 -dust no > ~{sample}_bl2seq_report.txt 2>error.log; then
echo 'blastn completed successfully' 1>&2
elif grep -q -F 'BLAST engine error: Warning: Sequence contains no data' error.log; then
# Copy the message to STDERR, and exit without an error status
cat error.log 1>&2
else
echo 'Unexpected error' 1>&2
cat error.log 1>&2
exit 1
fi
zcat ~{fastq} | sed -n '1~4p' | wc -l
set -euo pipefail
mkdir ~{sample}.SPAdes
if [ "~{readCount}" -gt "~{minReads}" ]; then
rnaspades.py --pe1-1 ~{fastq1} --pe1-2 ~{fastq2} -o ~{sample}.SPAdes
else
echo 'Not enough reads to run SPAdes genomic assembly.' 1>&2
fi
tar cf - ~{sample}.SPAdes | gzip --no-name > ~{sample}.SPAdes.tar.gz
For support, please file an issue on the Github project or send an email to gsi@oicr.on.ca .
Generated with generate-markdown-readme (https://github.com/oicr-gsi/gsi-wdl-tools/)