You signed in with another tab or window. Reload to refresh your session.You signed out in another tab or window. Reload to refresh your session.You switched accounts on another tab or window. Reload to refresh your session.Dismiss alert
In this tutorial, we'll use sourmash gather to analyze metagenomes using the LIN taxonomic framework.
Specifically, we will analyze plant metagenomes for the presence of Ralstonia solanacearum.
The goal is to see if we can correctly assign the sequence in each file to the correct phylogenetic group, distinguishing between pathogenic and non-pathogenic strains.
# create a new environment
mamba create -n sourmash -y -c conda-forge -c bioconda sourmash
then activate the conda environment:
conda activate sourmash
Victory conditions: your prompt should start with (sourmash)
and you should now be able to run sourmash and have it output usage information!!
Create a working subdirectory
Make a directory named smash_lin, change into it:
mkdir -p ~/smash_lin
cd ~/smash_lin
Now make a couple useful folders:
mkdir -p inputs
mkdir -p databases
Download relevant data
First, download a database and taxonomic information
Here, we know we are looking specifically for Ralstonia, so we can download a database
containing signatures of 32 Ralstonia genomes (pathogenic and not) and the corresponding taxonomic and lingroup information.
For cases where you do not yet know what organisms you may encounter, we provide pre-prepared databases for GTDB and GenBank here
Next, download pre-made sourmash signatures made from the input metagenomes
# download Sample-0 signature
curl -JLO https://osf.io/download/dvyt9/
# download Sample-II signature
curl -JLO https://osf.io/download/agwdu/
# download Sample-IV signature
curl -JLO https://osf.io/download/rngjq/
# move downloaded signatures to ./inputs
mv Sample*.zip ./inputs
# look at available input files
ls inputs
Look at the signatures
Let's start with the Sample-II sample
First, let's look at the metagenome signature.
By running sourmash sig fileinfo, we can see information on the signatures available within the zip file.
Here, you can see I've generated the metagenome signature with scaled=100 and built three ksizes, k=21, k=31andk=51`
Run:
sourmash sig fileinfo ./inputs/Sample-II.sc1000.zip
In the output, you should see:
** loading from './inputs/Sample-II.sc1000.zip'
path filetype: ZipFileLinearIndex
location: /Users/ntward/dib-lab/sandbox/smash_lin/inputs/Sample-II.sc1000.zip
is database? yes
has manifest? yes
num signatures: 3
** examining manifest...
total hashes: 105825
summary of sketches:
1 sketches with DNA, k=21, scaled=1000, abund 33335 total hashes
1 sketches with DNA, k=31, scaled=1000, abund 35516 total hashes
1 sketches with DNA, k=51, scaled=1000, abund 36974 total hashes
We can also look at the database
Here, you can see I've generated the database with scaled=1000 and built three ksizes, k=21, k=31 and k=51
Run:
sourmash sig fileinfo ./databases/ralstonia.zip
In the output, you should see:
** loading from './databases/ralstonia.zip'
path filetype: ZipFileLinearIndex
location: /Users/ntward/dib-lab/sandbox/smash_lin/databases/ralstonia.zip
is database? yes
has manifest? yes
num signatures: 96
** examining manifest...
total hashes: 524340
summary of sketches:
32 sketches with DNA, k=21, scaled=1000 174967 total hashes
32 sketches with DNA, k=51, scaled=1000 174975 total hashes
32 sketches with DNA, k=31, scaled=1000 174398 total hashes
There's a lot of things to digest in this output but the two main ones are:
there are 32 genomes represented in this database, each of which are sketched at k=21,k=31,k=51
this database represents ~524 million k-mers (multiply number of hashes by the scaled number)
selecting specified query k=31
loaded query: Sample-II... (k=31, DNA)
--
loaded 96 total signatures from 1 locations.
after selecting signatures compatible with search, 32 remain.
Starting prefetch sweep across databases.
Prefetch found 31 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
1.3 Mbp 3.9% 26.6% 1.2 GCF_001373295.1 Ralstonia solanacearum RS2
found less than 50.0 kbp in common. => exiting
found 1 matches total;
the recovered matches hit 3.9% of the abundance-weighted query.
the recovered matches hit 3.6% of the query k-mers (unweighted).
The first step of gather ("prefetch") found all potential matches with at least 50kb matching sequence (31 of 32 total database genomes). Then, the greedy algorithm narrowed this to a single best match, GCF_001373295.1 which shared an estimated 1.3 Mbp with the metagenome (~3.9% of the total query dataset). We can visualize this by looking at a venn diagram of the shared k-mers between metagenome sample and the top match. The yellow intersection represend <4% of the metagenome and ~26.6% of the Ralstonia RS2 reference genome. This small match percentage is expected, though, since the dataset is a simulated plant metagenome with an in silico Ralstonia spike-in, and we are just searching for Ralstonia here.
Add taxonomic information and summarize up lingroups
sourmash gather finds the smallest set of reference genomes that contains all the known information (k-mers) in the metagenome. In most cases, gather will find many metagenome matches. Here, we're only looking for Ralstonia matches and we only have a single gather result. Regardless, let's use sourmash tax metagenome to add taxonomic information and see if we've correctly assigned the pathogenic sequence.
First, let's look at the relevant taxonomy files.
These commands will show the first few lines of each file. If you prefer, you can look at a more human-friendly view by opening the files in a spreadsheet program.
Now, run sourmash tax metagenome to integrate taxonomic information into gather results
Using the gather output we generated above, we can integrate taxonomic information and summarize up "ranks" (lin positions). We can produce several different types of outputs, including a lingroup report.
lingroup format summarizes the taxonomic information at each lingroup, and produces a report with 4 columns:
name (from lingroups file)
lin (from lingroups file)
percent_containment - total % of the file matched to this lingroup
num_bp_contained - estimated number of bp matched to this lingroup
Since sourmash assigns all k-mers to individual genomes, no reads/base pairs are "assigned" to higher taxonomic ranks or lingroups (as with Kraken-style LCA). Here, "percent_containment" and "num_bp_contained" is calculated by summarizing the assignments made to all genomes in a lingroup. This is akin to the "contained" information in Kraken-style reports.
Trying to read LIN taxonomy assignments.
loaded 1 gather results from 'Sample-II.k31.gather.csv'.
loaded results for 1 queries from 1 gather CSVs
Read 20 lingroup rows and found 20 distinct lingroup prefixes.
Here, the most specific lingroup we assign to is A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1;, which means this is in phylotype IIB, sequevar 1. This is the USDA select agent!
:::
Now output the lingroup report to a file (instead of to the terminal)
use -o to provide an output basename for taxonomic output.
You should see saving 'lingroup' output to 'Sample-II.lingroup.tsv' in the output.
Optionally, write multiple output formats
You can use -F to specify additional output formats. Here, I've added csv_summary. Note that while the lingroup format will be generated automatically if you specify the --lingroup file, you can also specify it with -F lingroup if you want, as I've done here.
saving 'csv_summary' output to 'Sample-II.summarized.csv'.
saving 'lingroup' output to 'Sample-II.lingroup.txt'.
The csv_summary format is the full summary of this sample, e.g. the summary at each taxonomic rank (LIN position). It also includes an entry with the unclassified portion at each rank.
Note: Multiple output formats require the -o--output-base to be specified, as each must be written to a file.
Here's an abbreviated version of the gather results for Sample-II, with lingroup information added:
ksize
scaled
best overlap
gather match(es)
lingroup
lin
Sample-II
31
1000
1.3 Mbp
GCF_001373295.1
A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1
14;1;0;0;0;3;0;0;0;0;1;0;0;0;0;0;0
:::warning
Interlude - These samples were generated via read simulation from ralstonia genomes, and it looks like this one was created from the RS2 genome we are matching here. Let's try excluding this specific genome to see if we still find the same results without an exact database match. This should be more realistic.
We have a gather command-line option for just this situation, --exclude-db-pattern:
Starting prefetch sweep across databases.
Prefetch found 30 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
1.2 Mbp 3.9% 24.0% 1.2 GCF_002251655.1 Ralstonia solanacearum UW551
found less than 50.0 kbp in common. => exiting
found 1 matches total;
the recovered matches hit 3.9% of the abundance-weighted query.
the recovered matches hit 3.5% of the query k-mers (unweighted).
name lin percent_containment num_bp_contained
A_Total_reads;B_PhylII 14;1;0;0;0;3;0 3.91 1451000
A_Total_reads;B_PhylII;C_IIB 14;1;0;0;0;3;0;0 3.91 1451000
A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2 14;1;0;0;0;3;0;0;0;0;1;0;0;0;0 3.91 1451000
A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1 14;1;0;0;0;3;0;0;0;0;1;0;0;0;0;0;0 3.91 1451000
Without the exact genome the reads were generated from, we get a slightly smaller sequence overlap (1.2Mbp instead of 1.3Mbp). However, when we add the LINgroup information, we still find the right group, A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1, or Phylotype IIB, sequevar 1 (pathogenic).
:::
selecting specified query k=31
loaded query: Sample-0... (k=31, DNA)
--
loaded 96 total signatures from 1 locations.
after selecting signatures compatible with search, 32 remain.
Starting prefetch sweep across databases.
Prefetch found 0 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
found less than 50.0 kbp in common. => exiting
No matches found for --threshold-bp at 50.0 kbp.
gather found no sequence matches! Let's try lowering the detection threshold:
# use a 3kb detection threshold
sourmash gather inputs/Sample-0.sc100.zip databases/ralstonia.zip -k 31 --threshold-bp 3000 -o Sample-0.k31.gather.csv
This time, you should see:
selecting specified query k=31
loaded query: Sample-0... (k=31, DNA)
--
loaded 96 total signatures from 1 locations.
after selecting signatures compatible with search, 32 remain.
Starting prefetch sweep across databases.
Prefetch found 0 signatures with overlap >= 3.0 kbp.
Doing gather to generate minimum metagenome cover.
found less than 3.0 kbp in common. => exiting
No matches found for --threshold-bp at 3.0 kbp.
Which means we still didn't find anything! It turns out that Sample-0 is a control sample that does not have any Ralstonia.
:::info
Note: We typically recommend requiring matches to have 3 or more matching k-mers (threshold = 3 * scaled)
:::
Another option we won't pursue here is generating sketches for both the database and the queries at higher resolution (lower scaled, e.g. scaled=100). See more information on scaled and thresholds here.
Prefetch found 32 signatures with overlap >= 3.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
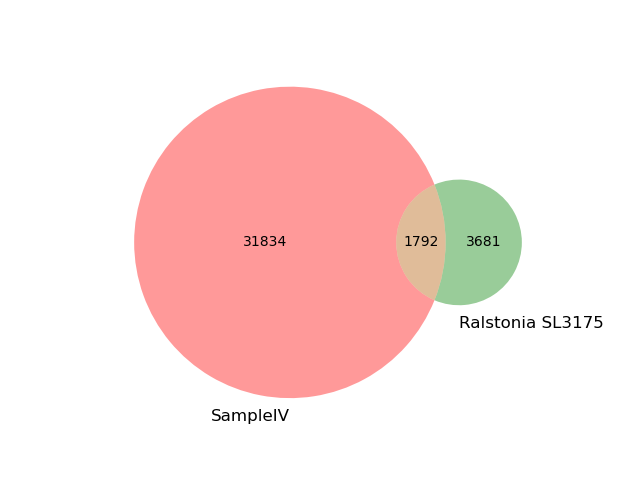
1.2 Mbp 3.7% 22.1% 1.2 GCF_003515185.1 Ralstonia solanacearum SL3175
found less than 3.0 kbp in common. => exiting
found 1 matches total;
the recovered matches hit 3.7% of the abundance-weighted query.
the recovered matches hit 3.4% of the query k-mers (unweighted).
We can look directly at the k-mer overlap between the SampleIV and this Ralstonia genome:
prefetch found 32 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
5.1 Mbp 17.8% 97.2% 57.5 GCF_002251605.2 Ralstonia solanacearum UW700
2.7 Mbp 0.3% 4.0% 21.2 GCF_000825845.1 Ralstonia solanacearum Grenada9_1
2.2 Mbp 0.0% 2.1% 5.0 GCF_001644815.1 Ralstonia solanacearum CFBP6783
0.8 Mbp 0.0% 1.6% 2.0 GCF_012062595.1 Ralstonia solanacearum Pe_13
4.9 Mbp 0.2% 1.2% 44.8 GCF_002251695.1 Ralstonia solanacearum K60
2.1 Mbp 0.0% 1.0% 1.4 GCF_000212635.3 Ralstonia solanacearum MolK2
found less than 50.0 kbp in common. => exiting
found 6 matches total;
the recovered matches hit 18.3% of the abundance-weighted query.
the recovered matches hit 0.6% of the query k-mers (unweighted).
The initial search (prefetch) found that all 32 genomes had shared sequence with our query. The minimum metagenome cover shows 6 genomes with non-overlapping matches. Nearly 18% of the abundance-weighted query matched to the GCF_002251605.2 Ralstonia solanacearum UW700 genome.
:::info
Just for fun, let's try adding a random tomato genome to the database, to see if we can match the host k-mers:
Starting prefetch sweep across databases.
Prefetch found 33 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
210.1 Mbp 19.8% 33.4% 1.5 GCF_000188115.5 Solanum lycopersicum
Heinz 1706
5.1 Mbp 17.8% 97.2% 57.5 GCF_002251605.2 Ralstonia solanacearum UW700
2.7 Mbp 0.3% 4.0% 21.2 GCF_000825845.1 Ralstonia solanacearum Grenada9_1
2.2 Mbp 0.0% 2.1% 5.0 GCF_001644815.1 Ralstonia solanacearum CFBP6783
0.8 Mbp 0.0% 1.6% 2.0 GCF_012062595.1 Ralstonia solanacearum Pe_13
4.9 Mbp 0.2% 1.2% 44.8 GCF_002251695.1 Ralstonia solanacearum K60
2.1 Mbp 0.0% 1.0% 1.4 GCF_000212635.3 Ralstonia solanacearum MolK2
found less than 50.0 kbp in common. => exiting
found 7 matches total;
the recovered matches hit 38.2% of the abundance-weighted query.
the recovered matches hit 21.7% of the query k-mers (unweighted).
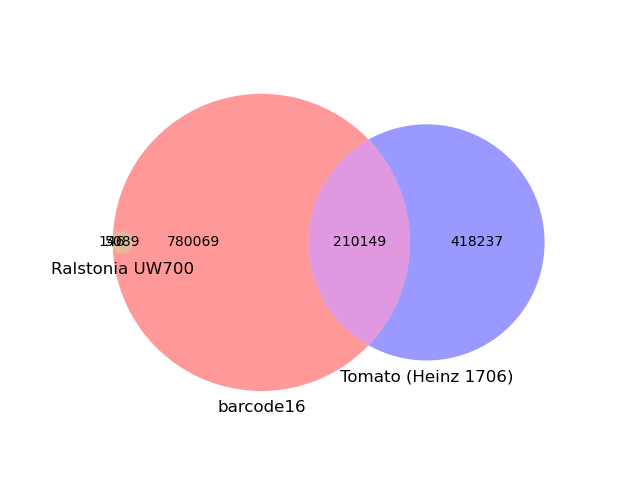
We can plot the k-mer overlap between this sample and the two top matches: Ralstonia UW700 and the Heinz 1706 tomato genome. Here, we see that while the Ralstonia k-mers are a small portion of the overall file (0.6%; small circle on the left), nearly the entire reference genome is present in the barcode16 sample (97.2%). This tomato genome, in constrast, shares a little less than half its content with the sample. It likely does not reflect the cultivar where bc16 was sampled from.
We can run tax metagenome with these results. However, since we don't have LIN or LINgroup information for the tomato genome, the results will only include the Ralstonia matches. Since the tomato genome did not share any k-mers with the Ralstonia genomes, there will be no impact on Ralstonia taxonomic assignment.
:::info
18% of the sample matched to A_Total_reads;B_PhylII;C_IIC, so we find that this samples is also in Phylotype IIC, which is not the USDA select agent and is not pathogenic.
:::
Summary and concluding thoughts
The LIN taxonomic framework may be useful distinguishing groups below the species level, and we can use LINs and LINgroups with sourmash tax. For low level matches, the gather greedy
approach can struggle. In cases where there is an identical % match between two reference genomes, the reported match is selected at random. We are working on ways to better warn users about places where this behavior occurs and welcome
feedback and suggestions on our issue tracker.
We typically recommend running at scaled=1000 (our default), as this works for most microbial use cases. However, for smaller samples and databases or for distinguishing between highly related genomes, you may want to run at higher resolution (lower scaled), e.g. scaled=100 or lower. Note, higher resolution signatures are larger and take longer to build and search. See more information on scaled and thresholds here.
Sourmash taxonomy can also be used with NCBI, GTDB, and ICTV taxonomies. For a walkthough using GTDB and sample-specific assemblies with an environmental metagenome, see here.
NOTE: We're in the process of upgrading sourmash commands for multithreading and faster processing. If you get comfortable with these commands and want to process more samples, please check out the branchwater plugin e.g. fastmultigather command for faster execution and to run multiple samples at once.
Post-workshop challenge: use the full GTDB reference database
While you can always create your own reference databases, as we've done here, one of sourmash's strengths is its ability to search large databases.
Here is a walkthrough for running the same analysis using the full GTDB reference database (rs214).
sourmash gather $query $database -k 31 -o $gather_csv_output
== This is sourmash version 4.8.9. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
selecting specified query k=31
loaded query: Sample-IV... (k=31, DNA)
--
loaded 402709 total signatures from 1 locations.
after selecting signatures compatible with search, 402709 remain.
Starting prefetch sweep across databases.
Prefetch found 350 signatures with overlap >= 50.0 kbp.
Doing gather to generate minimum metagenome cover.
overlap p_query p_match avg_abund
--------- ------- ------- ---------
1.2 Mbp 3.7% 22.1% 1.2 GCF_003515185.1 Ralstonia solanacearum strain=SL3175, ASM351518v1
found less than 50.0 kbp in common. => exiting
found 1 matches total;
the recovered matches hit 3.7% of the abundance-weighted query.
the recovered matches hit 3.4% of the query k-mers (unweighted).
Despite searching a much larger database which includes 526 Ralstonia genomes (350 of which had overlap >= 50.0 kbp), the gather results show that this sample is still most similar to GCF_003515185.1, which is in the Phylotype-IV LIN group.
The text was updated successfully, but these errors were encountered:
https://hackmd.io/191eqd9wQOSIPQyyjp5XUA
2024 ICPPB Sourmash LIN demo
Analyzing Metagenome Composition using the LIN taxonomic framework
Tessa Pierce Ward
July 2024
requires sourmash v4.8+
All materials for this workshop:
https://github.com/vinatzer-lab/ICPPB2024_workshop
This tutorial uses the
sourmash taxonomymodule, which was introduced via blog postand was recently shown to perfom well for taxonomic profiling of long (and short) reads in Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets, Portik et al., 2022.
In this tutorial, we'll use sourmash gather to analyze metagenomes using the LIN taxonomic framework.
Specifically, we will analyze plant metagenomes for the presence of Ralstonia solanacearum.
The goal is to see if we can correctly assign the sequence in each file to the correct phylogenetic group, distinguishing between pathogenic and non-pathogenic strains.
:::info
Simulated Samples: Ralstonia + tomato host:
Sample0- no RalstoniaSample-II- Ralstonia solanacearum, PhylIIBSampleIV- Ralstonia solanacearum, Phyl-IVAdditonal Sample (nanopore):
barcode16- ??Install sourmash
First, we need to install the software! We'll use conda/mamba to do this.
The below command installs sourmash.
Install the software:
then activate the conda environment:
Create a working subdirectory
Make a directory named
smash_lin, change into it:Now make a couple useful folders:
Download relevant data
First, download a database and taxonomic information
Here, we know we are looking specifically for Ralstonia, so we can download a database
containing signatures of 32 Ralstonia genomes (pathogenic and not) and the corresponding taxonomic and lingroup information.
Next, download pre-made sourmash signatures made from the input metagenomes
Look at the signatures
Let's start with the
Sample-IIsampleFirst, let's look at the metagenome signature.
By running
sourmash sig fileinfo, we can see information on the signatures available within the zip file.Here, you can see I've generated the metagenome signature with
scaled=100and built three ksizes,k=21, k=31andk=51`Run:
In the output, you should see:
We can also look at the database
Here, you can see I've generated the database with
scaled=1000and built three ksizes,k=21,k=31andk=51Run:
In the output, you should see:
There's a lot of things to digest in this output but the two main ones are:
Run sourmash gather using ksize 31
Now let's run
sourmash gatherto find the closest reference genome(s) in the database.If you want to read more about what sourmash is doing, please see Lightweight compositional analysis of metagenomes with FracMinHash and minimum metagenome covers, Irber et al., 2022.
Run:
You should see the following output:
The first step of gather ("prefetch") found all potential matches with at least 50kb matching sequence (31 of 32 total database genomes). Then, the greedy algorithm narrowed this to a single best match,
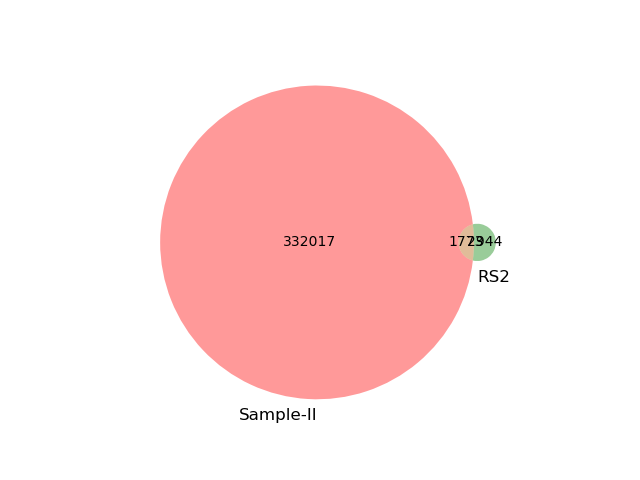
GCF_001373295.1which shared an estimated 1.3 Mbp with the metagenome (~3.9% of the total query dataset). We can visualize this by looking at a venn diagram of the shared k-mers between metagenome sample and the top match. The yellow intersection represend <4% of the metagenome and ~26.6% of the Ralstonia RS2 reference genome. This small match percentage is expected, though, since the dataset is a simulated plant metagenome with an in silico Ralstonia spike-in, and we are just searching forRalstoniahere.Add taxonomic information and summarize up lingroups
sourmash gatherfinds the smallest set of reference genomes that contains all the known information (k-mers) in the metagenome. In most cases,gatherwill find many metagenome matches. Here, we're only looking forRalstoniamatches and we only have a single gather result. Regardless, let's usesourmash tax metagenometo add taxonomic information and see if we've correctly assigned the pathogenic sequence.First, let's look at the relevant taxonomy files.
These commands will show the first few lines of each file. If you prefer, you can look at a more human-friendly view by opening the files in a spreadsheet program.
databases/ralstonia32.lin-taxonomy.csvlin(14;1;0;...) andaccession(GCF_00...)databases/ralstonia.lingroups.csvname,lin)Look at the taxonomy file:
You should see:
Now, let's look at the lingroups file
You should see:
Now, run
sourmash tax metagenometo integrate taxonomic information intogatherresultsUsing the
gatheroutput we generated above, we can integrate taxonomic information and summarize up "ranks" (lin positions). We can produce several different types of outputs, including alingroupreport.lingroupformat summarizes the taxonomic information at eachlingroup, and produces a report with 4 columns:name(from lingroups file)lin(from lingroups file)percent_containment- total % of the file matched to this lingroupnum_bp_contained- estimated number of bp matched to this lingroupRun
tax metagenome:You should see:
and the results:
:::info
Here, the most specific lingroup we assign to is
A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1;, which means this is in phylotype IIB, sequevar 1. This is the USDA select agent!:::
Now output the lingroup report to a file (instead of to the terminal)
use
-oto provide an output basename for taxonomic output.Optionally, write multiple output formats
You can use
-Fto specify additional output formats. Here, I've addedcsv_summary. Note that while thelingroupformat will be generated automatically if you specify the--lingroupfile, you can also specify it with-F lingroupif you want, as I've done here.Run:
You should see the following in the output:
The
csv_summaryformat is the full summary of this sample, e.g. the summary at each taxonomic rank (LIN position). It also includes an entry with theunclassifiedportion at each rank.Here's an abbreviated version of the
gatherresults forSample-II, with lingroup information added::::warning
Interlude - These samples were generated via read simulation from ralstonia genomes, and it looks like this one was created from the RS2 genome we are matching here. Let's try excluding this specific genome to see if we still find the same results without an exact database match. This should be more realistic.
We have a gather command-line option for just this situation,
--exclude-db-pattern:gather:
gather results:
tax:
tax results:
Without the exact genome the reads were generated from, we get a slightly smaller sequence overlap (1.2Mbp instead of 1.3Mbp). However, when we add the LINgroup information, we still find the right group,
A_Total_reads;B_PhylII;C_IIB;D_seq1&seq2;E_seq1, or Phylotype IIB, sequevar 1 (pathogenic).:::
Run the other samples
Now run with
Sample-0samplesourmash gather
Run:
You should see:
gather found no sequence matches! Let's try lowering the detection threshold:
This time, you should see:
Which means we still didn't find anything! It turns out that Sample-0 is a control sample that does not have any Ralstonia.
:::info
Note: We typically recommend requiring matches to have 3 or more matching k-mers (threshold =
3 * scaled):::
Another option we won't pursue here is generating sketches for both the database and the queries at higher resolution (lower scaled, e.g. scaled=100). See more information on scaled and thresholds here.
Run the last sample, Sample-IV
we see:
We can look directly at the k-mer overlap between the SampleIV and this Ralstonia genome:
run tax metagenome
LINgroup output file:
:::info
We find that this genome is in Phylotype IV, seq10 group (non-pathogenic).
:::
run the infected field sample ("barcode 16"; nanopore):
Download the signature
run gather
gather results:
The initial search (prefetch) found that all 32 genomes had shared sequence with our query. The minimum metagenome cover shows 6 genomes with non-overlapping matches. Nearly 18% of the abundance-weighted query matched to the GCF_002251605.2 Ralstonia solanacearum UW700 genome.
:::info
Just for fun, let's try adding a random tomato genome to the database, to see if we can match the host k-mers:
new results:
We can plot the k-mer overlap between this sample and the two top matches: Ralstonia UW700 and the Heinz 1706 tomato genome. Here, we see that while the Ralstonia k-mers are a small portion of the overall file (0.6%; small circle on the left), nearly the entire reference genome is present in the barcode16 sample (97.2%). This tomato genome, in constrast, shares a little less than half its content with the sample. It likely does not reflect the cultivar where bc16 was sampled from.
run tax metagenome
We can run tax metagenome with these results. However, since we don't have LIN or LINgroup information for the tomato genome, the results will only include the Ralstonia matches. Since the tomato genome did not share any k-mers with the Ralstonia genomes, there will be no impact on Ralstonia taxonomic assignment.
LINgroup results:
:::info
18% of the sample matched to A_Total_reads;B_PhylII;C_IIC, so we find that this samples is also in Phylotype IIC, which is not the USDA select agent and is not pathogenic.
:::
Summary and concluding thoughts
The LIN taxonomic framework may be useful distinguishing groups below the species level, and we can use LINs and LINgroups with
sourmash tax. For low level matches, the gather greedyapproach can struggle. In cases where there is an identical % match between two reference genomes, the reported match is selected at random. We are working on ways to better warn users about places where this behavior occurs and welcome
feedback and suggestions on our issue tracker.
We typically recommend running at
scaled=1000(our default), as this works for most microbial use cases. However, for smaller samples and databases or for distinguishing between highly related genomes, you may want to run at higher resolution (lower scaled), e.g. scaled=100 or lower. Note, higher resolution signatures are larger and take longer to build and search. See more information on scaled and thresholds here.Sourmash taxonomy can also be used with NCBI, GTDB, and ICTV taxonomies. For a walkthough using GTDB and sample-specific assemblies with an environmental metagenome, see here.
NOTE: We're in the process of upgrading sourmash commands for multithreading and faster processing. If you get comfortable with these commands and want to process more samples, please check out the branchwater plugin e.g.
fastmultigathercommand for faster execution and to run multiple samples at once.Post-workshop challenge: use the full GTDB reference database
While you can always create your own reference databases, as we've done here, one of sourmash's strengths is its ability to search large databases.
Here is a walkthrough for running the same analysis using the full GTDB reference database (rs214).
For all available databases, see sourmash prepared databases.
output:
Despite searching a much larger database which includes 526 Ralstonia genomes (350 of which had overlap >= 50.0 kbp), the gather results show that this sample is still most similar to
GCF_003515185.1, which is in the Phylotype-IV LIN group.The text was updated successfully, but these errors were encountered: